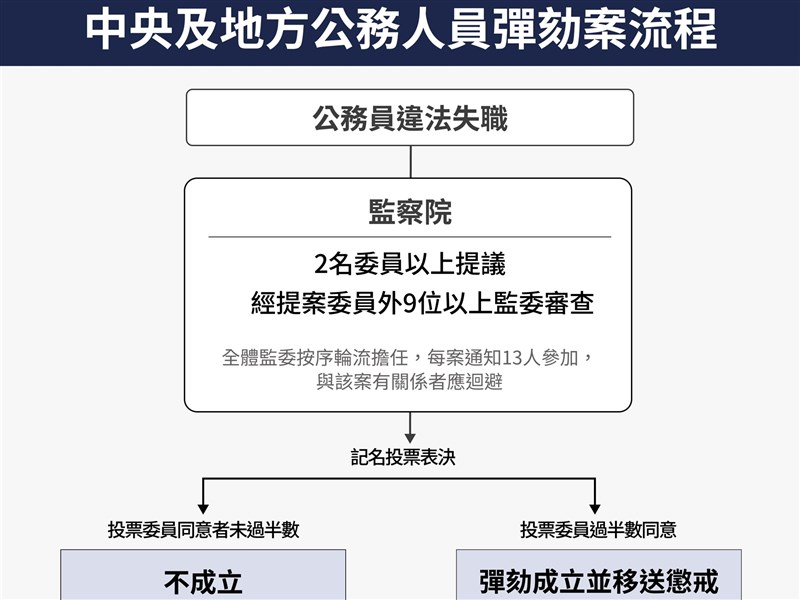






聯亞藥生物相似藥將申請台灣藥證 今年擬股票上市
(中央社記者韓婷婷台北20日電)興櫃生技廠聯亞藥今天公布重組人類紅血球生成素生物相性藥UB-851第三期人體臨床試驗結果,期末數據分析符合預期,後續將送件申請產品上市;另外,聯亞藥預計最快今年中將送件申請股票掛牌上市。
聯亞藥召開重大訊息記者會,公布旗下研發的重組人類紅血球生成素生物相性藥品UB-851第三期人體臨床試驗,已完成期末數據分析,試驗結果符合預期,後續將送件申請產品上市查驗登記審查,並申請台灣藥證。
至於集團聯亞生技研發的2019冠狀病毒疾病(COVID-19)疫苗最新進度,聯亞藥董事長陳啟祥指出,之前雖沒通過緊急使用授權(EUA),但仍有1500名受試者參與第3劑的延伸試驗,預計將等數據出爐後才會有進一步動作。
聯亞藥執行副總經理兼發言人范瀛云表示,此一生物相似藥已完成三期臨床,並符合預期,預計將是最優先可望上市的自有研發藥。
另外,范瀛云說,聯亞藥也準備送件申請股票掛牌上市,預計最快在今年年中送件;因應首次公開募股(IPO)財結構調整,聯亞藥日前已公告辦理現金增資,預計發行600萬股,每股發行價格50元,現金增資認股基準日為2月15日。
聯亞藥開發的紅血球生成素UB-851,適應症為治療腎性貧血症。三期臨床試驗採多劑量、隨機雙盲及平行對照設計,以比較UB-851產品與經衛生福利部食品藥物管理署(TFDA)核准參考藥物J&J所開發Eprex在療效、安全性與免疫原性的相似性。
臨床試驗實際受試者人數為204人,以2:1比例分配在UB-851與Eprex組,試驗期間為24週比較UB-851與Eprex使用安全性、免疫原性與維持目標血紅素療效相等性,及UB-851延續試驗期間至52週長期使用安全性與免疫原性。

聯亞藥指出,期末分析結果顯示,在臨床安全性方面,UB-851組別無抗紅血球生成素抗體產生,顯示UB-851長期使用安全性與無免疫原性。
聯亞藥表示,UB-851已完成第三期人體臨床試驗及資料分析數據,將於試驗細部分析報告及應主管機關要求的藥品製造等文件整理完成後,後續將向衛福部食藥署(TFDA)申請生物相似性藥品上市許可查驗登記。(編輯:康世人)1110120
本網站之文字、圖片及影音,非經授權,不得轉載、公開播送或公開傳輸及利用。